
The Sanderson Group Webpages
Department of Chemistry
Durham University, Durham, UK
Synthetic Antimicrobial Peptides
An increasing number of peptides that posess antimicrobial activity as a result of membrane-lytic activity are being characterised. These peptides have been isolated from a wide range of sources, including plants, mammals, insects, reptiles, fungi and bacteria. Perhaps the most celebrated examples were isolated from the skin of crocodiles following an investigation into the mechanisms by which these reptiles are able to survive in relatively hostile microbial environments without succumbing to infection. In humans, antimicrobial peptides have been isolated from a number of bodily fluids, including saliva and blood plasma, and are implicated as an intrinsic part of the immune system.
Clinial Applications of Antimicrobial Peptides
To date, the number of bactericidal peptides that are used in clinical treatments is small. Those that have found application, such as gramicidin, are used topically to treat skin and wound infections, as internal administration produces strong (anaphylactic) immune responses. In order to address this problem, we are attempting to design new peptides that use non-natural amino acids of low immunogenic activity. At the same time, we are refining the selectivity of the peptides to enable them to act upon a narrow range of bacterial species, as this will ultimately reduce the toxicity to mammalian cells and reduce the spread of resistant bacterial strains.
Designed Peptides
In the initial stages of our work,1 we performed bioinformatics excercises on prokaryotic β-barrel membrane proteins to enable us to define the key structural features of these proteins essential for the formation of stable transmembrane structures. These proteins form particularly stable channels in the outer membrane of Gram negative bacteria, and were therefore an obvious target for our research. Using our initial results, we designed a number of peptides, such as cyclic peptide 1, that we predicted would assemble in liposomal membranes to form ion channels of suitable diameter to enable the escape of the liposomal contents.
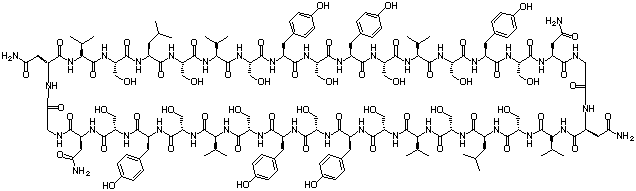
1
A small library of these peptides was synthesised using solid-phase methodology and screened against a preparation of liposomes containing entrapped carboxyfluorescein. Carboxyfluorescein exhibits fluorescence self-quenching at high concentrations (> 30 mM), but is strongly fluorescent at low concentrations (mM range), so release of the fluorophore entrapped in liposomes at high concentration into the surrounding medium produces an increase in fluorescence intensity that can be monitored using conventional fluorescence spectroscopy techniques. Screening of our initial library identified two peptides that provoked the release of entrapped carboxyfluorescein from liposomes at peptide:lipid ratios as low as 1:2500. Experiments using laser correlation spectroscopy on the liposomes in the absence and presence of the peptides produced essentially idenitcal results, consistent with the formation of peptide channels rather than complete disruption of the liposomal membrane. We are now performing a detailed characterisation of these peptides and extending our peptide libraries in the search for other similar peptides of high potency.
Kinetics of Membrane Binding
Cyclic peptides such as 1 have proved to be useful tools for studying the kinetics of peptide association with membranes by LD spectroscopy.2 Peptides with tyrosine adjacent to the β-turn exhibit 2-stage binding to PC membranes, with a rapid initial phase (seconds) followed by a slower phase (minutes to hours). Replacing the two tyrosines with tryptophans produces a 3-stage binding process. In all cases, the rapid initial phase has kinetics that match those of marker release.
References
- "The Design, Synthesis and Characterisation of Channel-Forming Peptides", John M. Sanderson and Sarah Yazdani, Chem. Commun., 2002, 1154–1155.
- "Peptide Adsorption to Lipid Bilayers: Slow Processes Revealed by Linear Dichroism Spectroscopy", Sue M. Ennaceur, Matthew R. Hicks, Catherine J. Pridmore, Tim R. Dafforn, Alison Rodger and John M. Sanderson, Biophys. J., 2009, 96, 1399–1407.